DNA SCIENCE
WEEK 2
Objectives:
Extraction, Ligation and
Transformation
Introduction:
This week, the desired DNA was
retrieved from the previous electrophoresis agarose gel and purified. To
insert our desired DNA fragment into the bacteria plasmids, ligation was
first performed to join the complementary sticky ends (that were cut by
the restriction enzymes last week) onto the vectors. Subsequently, cells
containing DH5a
strains were transformed to allow the bacteria plasmids to take up the
modified vectors.
Methods:
A1) Extraction (DNA-insert)
1. Desired DNA bands on the gel
are cut out and isolated in eppendorf tubes. The weight of the gel is
noted to be about 1.61g.
2. Isolated gel is dissolved in
Buffer QG from the Qiagen Gel Extraction Kit. Amount of buffer to
be added is 3 times the weight of the gel (i.e. 4.83 ml). Dissolution is
allowed to occur at 50degC for about 5-10 minutes.
3. Proceeding gel extraction
steps were carried out as per the
Qiagen Gel Extraction Assay.
Briefly, isopropanol is added to increase the yield of the desired DNA
fragments. Subsequently, it is then centrifuged @ 13,200g for 1 min to
retain the DNA in the column. This process is repeated until the entire
volume is depleted. To further remove the agarose gel, Buffer PE is
added and centrifugation is repeated.
4. To elute the DNA, Buffer EB is
added and again centrifuged. The final DNA was collected in the
filtrate.
A2) Extraction (PCR-vector)
1. HindIII and KpnI were added to
the previous PCR products to obtain “sticky ends”.


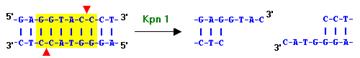
2. The digested fragments were
purified using the Qiagen PCR Purification kit.
Briefly, 5 volumes of Buffer PB
is added to the PCR fragments and centrifuged for 1 min to bind the DNA
to the column. Buffer PE is added to wash the PCR extracted, and again
centrifuged for 1 min @ 13,200g. To elute the desired PCR product,
buffer EB is added, centrifuged and the final PCR product is collected
in the filtrate.
B) Ligation
1. DNA ligation is performed
according to the Roche Rapid DNA Ligation Kit
protocol.
Briefly, mixture of various
proportions* of vector and insert were added to conc. DNA Dilution
Buffer to a final concentration of 10µL. 10µL of T4 DNA ligation buffer
is then added and mixed thoroughly. Finally 1 µL of T4 DNA ligase is
added and the mixture is incubated at 15-25degC for 5 mins.
|
Ratio |
Vector (µL) |
Insert (µL) |
DNA dilute buffer (µL) |
|
1:3 |
3.6 |
2.8 |
3.6 |
|
1:1 |
1.2 |
2.8 |
6.0 |
|
3:1 |
0.4 |
2.8 |
6.8 |
|
No
insert |
1.2 |
0 |
8.8 |
|
No
vector |
0 |
2.8 |
7.2 |
* Various
proportions of vector and insert
2. The same plasmid was treated
with a killer cut to ligate the center of the LacZ DNA sequence to
prevent religation and so that cells containing this killer cut do not
produce and eventually die. This serves as a control for each ratio of
vector and insert.
C1) Transformation
1. Electrocompetent E. Coli (Escherichia
coli) cells were grown
to an OD of 0.6, washed with glycerol and allowed to incubate in
LB for 30 mins before electroporation. This is to allow the cells to
develop some anti-biotic resistance and not lose their plasmids later
during transformation.
2.
Electroporation is the
process where cells are allowed to uptake a foreign DNA by means of
stressing the cells via an electric field. This process was carried out
as per the same Roche Rapid DNA Ligation Kit protocol. Briefly,
100 µL of Binding Buffer is added to the ligation reaction mixture
obtained in step 1 and centrifuged. 500 µL of wash buffer is then added
to the upper reservoir of the filter tube and again centrifuged. This
process is repeated twice. 100 µL of sterile double distilled water is
finally added, centrifuged and the flowthrough is retained. Roughly 10
µL of the flowthrough is then added to the electrocompetent cells in
Step 1 and then subjected to electroporation. The settings for
electroporation were: 2.5 kV, 25 MF, 200ohm.
3. The electroporated cells were
then plated and grown @ 37degC for 12-15 hours. The following were added
in the agar where cells were grown:
-
Kanamycin
- Antibiotic (cells without the Kanamycin resistance will die, i.e.
cells which did not have our DNA sequence in their plasmids)
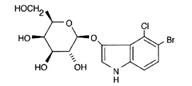
-
X-gal – (galactose
sugar with a glycosidic linkage to a chromophor (colored) molecule.
In the presence of the enzyme beta-galactosidase the glycosidic link
is hydrolyzed (broken by the addition of water and a blue color
results)
-
ATC
(anhydrotetracycline)
– Inducer (cells with a TEC (tetracycline)
repressor sitting on the promoter site require ATC to turn on the
LacZ gene; otherwise, X-gal will not turn blue)
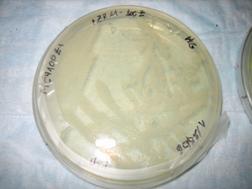
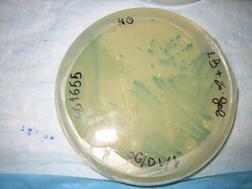
Results
Shown below, was the Petri dish
containing no X-gal. As expected, no blue color can be seen.
In the other type of cells
containing the strain DH5a-Z1/MC4100Z1,
the TEC repressor is present, and hence only with the addition of ATC
inducer, can Lac Z be expressed. Hence as shown in the dish on the left
(ATC absent), virtually no blue color could be detected. This is
compared to the vivid blue color found in the dish on the right, with
the presence of ATC.
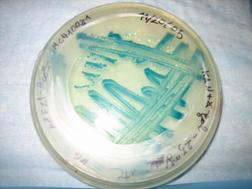
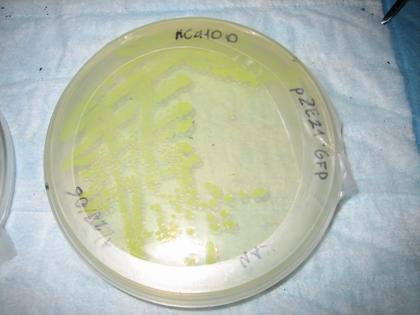
Also plated were the pZE21-GFP
cells (for fun) – fluorescence was observed as expected.